
TL;DR:
- Gene variant analysis identifies DNA differences that influence health by using tools like Ensembl VEP and the ACMG classification system. It involves three steps: calling variants, annotating them, and interpreting their impact within the clinical context. Variants are classified into five categories, with ongoing updates based on new evidence and research.
Gene variant analysis is the systematic process of examining DNA sequence variations to determine their potential impact on health and disease. Clinicians and researchers use it to identify changes in a person's genetic code, place those changes in biological context, and decide whether they raise or lower disease risk. Tools like Ensembl VEP and the ACMG classification system have made this process more consistent and reproducible. For patients, parents, and health-conscious consumers, understanding what gene variant analysis is and how it works is the first step toward making informed decisions about genetic testing.
What is gene variant analysis and why does it matter?
Gene variant analysis is the end-to-end process of detecting, annotating, and interpreting differences in DNA sequence that may affect a person's health. Every human genome contains millions of variants compared to a reference sequence. Most are harmless. A small fraction drives disease risk, influences drug response, or shapes inherited conditions like hereditary breast cancer linked to BRCA1 and BRCA2 mutations.

The importance of gene variants extends well beyond cancer. Variants in genes like CYP2D6 affect how the liver metabolizes common medications, including antidepressants and blood thinners. Variants in MLH1 and MSH2 raise the risk of Lynch syndrome, a hereditary colorectal cancer condition. Identifying these variants early gives patients and physicians the chance to act before symptoms appear, shifting care from reactive treatment to prevention.
Gene mutation analysis and gene variant analysis are often used interchangeably in consumer contexts, but clinicians prefer "variant" because not every genetic difference is a mutation in the disease-causing sense. The word "variant" is neutral and reflects the full spectrum from clearly harmful to clearly harmless.
What are the main steps involved in gene variant analysis?
The variant analysis workflow runs through three sequential phases: variant calling, variant annotation, and variant interpretation. Each phase builds on the last, and errors in any one phase affect the quality of the final clinical report.
1. Variant calling
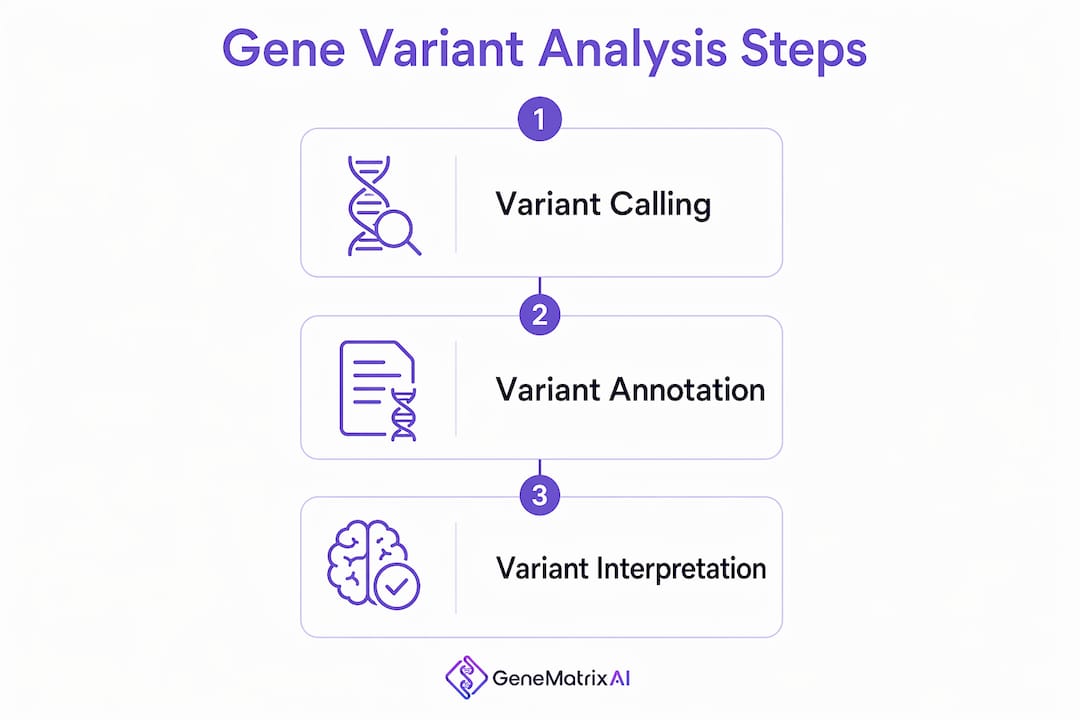
Variant calling identifies raw genetic changes from sequencing data. Common calling tools include GATK HaplotypeCaller, MuTect2, and DRAGEN. This phase is the most computationally demanding. Parameter tuning matters here because poorly calibrated settings produce false positives (flagging harmless variants as suspicious) or false negatives (missing real disease-causing changes).
2. Variant annotation
Raw genomic data files, called VCF files, are coordinate lists that no clinician can read directly. Annotation translates these files into clinically meaningful reports by cross-referencing external databases. Tools like Ensembl VEP, SnpEff, and ANNOVAR attach biological and clinical context to each variant. Databases like ClinVar and gnomAD supply population frequency data and previously documented clinical significance.
3. Variant interpretation
Interpretation is where science meets clinical judgment. Analysts apply the ACMG framework to classify each variant into one of five tiers. This step integrates computational predictions, published literature, and patient-specific data including symptoms and family history.
Pro Tip: Ask your genetic testing provider which annotation databases their pipeline references. A report built on ClinVar and gnomAD carries more clinical weight than one using a single proprietary database.
How are gene variants classified and what does each classification mean?
The ACMG five-tier classification system is the global standard for clinical variant classification as of 2026. The framework uses 28 criteria grouped by evidence type, including population frequency, computational predictions, functional study results, family segregation data, and whether the variant occurred spontaneously (de novo) rather than being inherited.
| Classification | Meaning | Clinical action |
|---|---|---|
| Pathogenic | Strong evidence the variant causes disease | Immediate clinical follow-up |
| Likely Pathogenic | Evidence leans toward disease-causing | Clinical follow-up recommended |
| Variant of Uncertain Significance (VUS) | Insufficient evidence to classify | Monitor; reclassify as evidence grows |
| Likely Benign | Evidence leans toward harmless | Typically no clinical action |
| Benign | Strong evidence the variant is harmless | No clinical action needed |
The VUS category deserves special attention. A VUS is not a diagnosis. It is a clinical placeholder subject to reclassification as new research emerges or as more family members are tested. Patients who receive a VUS result should stay in contact with their genetic counselor and expect periodic updates.
Pro Tip: If your report includes a VUS, ask your provider to enroll you in a variant registry. Registries like ClinVar aggregate data from thousands of patients, which accelerates reclassification.
The classification of a variant can change over time. A variant labeled Likely Benign today may be reclassified as Pathogenic in three years if new functional studies reveal disease-causing effects. This dynamic nature of gene variant significance is one reason ongoing genetic counseling matters as much as the initial test.
What tools and databases are essential for gene variant annotation?
Effective gene variant interpretation depends on combining multiple tools rather than relying on any single platform. Each tool has strengths and blind spots, and cross-referencing results improves confidence in the final classification.
- Ensembl VEP (Variant Effect Predictor): Developed by the European Bioinformatics Institute, VEP predicts the functional consequences of variants across the genome. It integrates data from dozens of databases and is widely used in both research and clinical pipelines.
- SnpEff: Annotates variants with their predicted effects on genes and proteins. It is particularly useful for identifying variants that disrupt protein-coding sequences.
- ANNOVAR: Annotates variants using population frequency data from gnomAD and clinical significance data from ClinVar. It handles large variant sets efficiently.
- GATK Funcotator: Part of the GATK toolkit from the Broad Institute, Funcotator adds functional annotation directly within the GATK variant calling pipeline, reducing the number of separate processing steps.
- ClinVar: A public database maintained by the National Center for Biotechnology Information (NCBI) that archives relationships between genetic variants and human health. It is the primary reference for known pathogenic variants.
- gnomAD: The Genome Aggregation Database aggregates sequencing data from over 125,000 exomes and 15,000 whole genomes. High population frequency in gnomAD is strong evidence that a variant is benign.
Tools like ACVI-Med take this further by helping non-expert analysts filter and prioritize variants according to ACMG guidelines through an interface designed for clinical settings rather than bioinformatics labs. That kind of phenotype-aware filtering is what separates a usable clinical report from a raw data dump.
Computational predictions are powerful but not definitive. Different prediction algorithms frequently disagree on whether a variant damages protein function. That disagreement is a signal, not a failure. It means the variant needs functional validation before a clinical decision is made.
Why does expert interpretation matter even with advanced tools?
Automated tools act as decision support, not final verdicts. Algorithms require human clinical oversight because computational predictions vary between models and cannot account for the full clinical picture a patient presents.
"Variant interpretation is not purely computational. Patient symptoms, family history, and published literature all influence classification." — ACMG-aligned clinical practice guidance
A patient with a BRCA2 variant classified as Likely Pathogenic by an algorithm may have no family history of breast or ovarian cancer. That context does not change the classification, but it shapes how a clinician communicates risk and plans surveillance. Conversely, a patient with a strong family history and a VUS result may warrant closer monitoring than the classification alone suggests.
Functional validation experiments are the gold standard when computational predictions conflict. These lab-based assessments measure protein stability, enzyme activity, or cellular signaling effects to confirm whether a variant actually disrupts biological function. Functional data can move a VUS to Pathogenic or Benign, resolving clinical uncertainty. Proper lab contamination prevention during these experiments is critical because contaminated samples produce unreliable results that can mislead classification.
The best clinical genomics providers build human review into every stage of their pipeline. Automated tools accelerate the process. Clinicians and genetic counselors make it trustworthy.
Key Takeaways
Gene variant analysis converts raw DNA sequencing data into clinically actionable health insights through a three-phase workflow of calling, annotation, and expert interpretation guided by the ACMG five-tier classification system.
| Point | Details |
|---|---|
| Three-phase workflow | Variant calling, annotation, and interpretation must all be done accurately for reliable results. |
| ACMG five-tier system | Classifications range from Pathogenic to Benign, with VUS as a dynamic placeholder requiring ongoing review. |
| Tools work together | Combining Ensembl VEP, ClinVar, and gnomAD produces more reliable annotations than any single tool alone. |
| Human oversight is required | Automated algorithms support clinical decisions but cannot replace expert evaluation of patient-specific data. |
| VUS is not a diagnosis | A Variant of Uncertain Significance may be reclassified as new evidence emerges, making follow-up care essential. |
Gene variant results are more of a conversation than a verdict
I have spent years watching patients receive genetic test results and immediately search for a yes or no answer. The biology rarely cooperates. A Pathogenic result on a BRCA1 variant does not mean cancer is inevitable. A VUS does not mean something is wrong. What it means is that the science is still catching up.
The part that most articles skip is how iterative this process actually is. A result you receive today is based on the evidence available today. As more people share their genomic data through registries and research programs, classifications shift. I have seen VUS results reclassified to Likely Benign within two years of an initial report. That is not a failure of the original analysis. That is the system working as designed.
My strongest advice for anyone navigating a genetic test result: do not try to interpret it alone. Genetic counselors exist specifically to translate these classifications into language that fits your personal and family history. They also track reclassifications and reach out when your result changes. That ongoing relationship is worth more than the initial report.
The 2026 precision medicine landscape is moving toward AI-assisted interpretation that flags reclassification candidates automatically. That is a meaningful improvement. But the human conversation that follows the report is still where the real value lives.
— Tarek
How Genematrix supports accurate gene variant analysis
Genematrix is a Chicago-based, CLIA-certified biotechnology company that applies AI-driven analysis to hereditary cancer screening, pharmacogenomics, and personalized wellness. Its GeneMatrixAI platform is trained on 500,000+ genetic profiles and delivers reports within 72 hours.
Genematrix offers specialized testing modules including GeneCancer (BRCA1/BRCA2, Lynch syndrome), GenePGx (drug-gene interactions), GeneMind (psychiatric pharmacogenomics), GeneBaby (pediatric genetics), and GeneDiet (nutrigenomics). Each report follows ACMG-aligned classification standards with clinical review built into the workflow. Patients who want to understand the science behind their results can visit the Genematrix science page for details on the technology, lab certifications, and research standards. The GeneMatrixAI app makes those insights accessible on iOS and Android for ongoing health management.
FAQ
What is gene variant analysis in simple terms?
Gene variant analysis is the process of identifying differences in a person's DNA, placing those differences in biological context, and determining whether they affect health or disease risk.
What does a VUS result mean for my health?
A Variant of Uncertain Significance means there is not yet enough evidence to classify the variant as harmful or harmless. It is a placeholder, not a diagnosis, and may be reclassified as new research emerges.
How are gene variants classified?
The ACMG five-tier system classifies variants as Pathogenic, Likely Pathogenic, Variant of Uncertain Significance, Likely Benign, or Benign based on 28 evidence criteria including population frequency and functional study data.
What tools are used to analyze gene variants?
Common tools include GATK HaplotypeCaller and MuTect2 for variant calling, and Ensembl VEP, SnpEff, and ANNOVAR for annotation, all cross-referenced against databases like ClinVar and gnomAD.
Can a gene variant classification change over time?
Yes. Classifications, especially VUS results, are updated as new clinical data, functional studies, and population frequency information become available through ongoing research and variant registries.